본 시리즈에서는 특별히 ChIP-seq data를 분석하는 방법에 대해 초점을 맞추어 살펴볼 것임.

ChIP-seq은 위와 같은 의미를 가지고 있으며, 이 중 특별히 이번 chapter에서는 기본적인 sequencing 관련 사항들에 대해 공부해볼 것임.
DNA sequencing은 nucleic acid sequence를 결정하는 과정이며, 구체적으로 DNA 내에 있는 nucleotide들의 순서를 파악하는 과정임.

DNA sequencing에 대한 역사는 위와 같음. 보면 일단 1963년에 Watson과 Crick에 의해 DNA의 구조가 밝혀짐. 이후 1965년에 우선 tRNA의 sequence가 최초로 먼저 sequencing되었는데, 그도 그럴 것이 이 당시에는 RNA가 genetic material이라 생각하는 사람이 많았고 그러다 보니 연구하는 과정에서 먼저 sequencing에 성공한 것임. 이후 1977년에 Fredric Sanger에 의해 chain-termination method가 개발되었으며, 이후 1986년에 최초의 automated DNA sequencing method가 개발됨. 이후 sequencing platform의 지속적인 발전이 이루어져 옴.

위 그림은 각종 sequencing method들을 비교해서 나타내주고 있는데, 이 때 x축은 한 가닥의 서열을 넣어줬을 때 읽을 수 있는 read length를 의미하며, y축은 throughput을 의미함.
보면 first-generation sequencing method인 Sanger sequencing과 비교했을 때, 나머지 NGS method들의 throughput이 훨씬 더 높음을 알 수 있음. 이들의 throughput이 높을 수 있는 이유는 위 그림 오른쪽 상에 나온 Flowcell을 이용해 massively parallel하게 sequencing을 하기 때문인데, 구체적으로 하나하나의 lane에 parallel하게 DNA들을 immobilize시켜놓은 상태에서 동시에 sequencing을 수행함.

위 그림을 보면 알 수 있는것처럼, sequencing 기술이 발전함에 따라 sequencing cost가 급감함. 현재는 single human genome을 sequencing하기 위해 단돈 400달러만 주면 됨.

한편 위 그림상에 나타나 있는 Illumina Novaseq과 BGI T7은 대표적인 NGS 장비임. 이 중 Illumina 장비를 이용하면 48명의 human whole genome을 40X로 sequencing하는데에 2일이면 충분함. 한편 BGI 장비를 이용하면 60명의 human whole genome을 30X로 sequencing하는데에 1일이면 충분함.
다음으로 sequencing output에 대해 조금 더 알아보자.

일단 이에 대한 이해를 위해서는 위와 같은 정보들에 대해 이해할 필요가 있음. 이 때 PE라고 적인 것은 paired end를 의미하며, paired end란 위 그림 오른쪽과 같이 양쪽에서 서열을 읽어나가는 sequencing system을 의미함. (이 때 화살표로 나타내진 두 부분이 실제로 읽히는 부분이며, 그림상에서 이 부분 각각에 대한 2개 종류의 read가 나올 것임) 그리고 data output은 output으로 나오는 read 서열 전체의 output 길이 총합을 의미함.
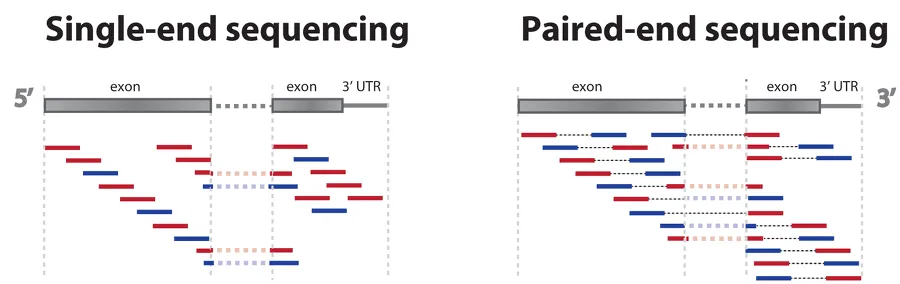
위 그림은 single-end sequencing과 paired-end sequencing 간의 차이점을 보여주고 있음. 우선 single-end sequencing이란 한쪽 방향씩으로만 읽어나가는 sequencing 기법이며, paired end sequencing은 앞서 말했던 것처럼 양쪽 방향에서 읽어나가는 sequencing 기법을 의미함. 이 중 paired-end sequencing을 할 시 양쪽의 조금씩을 sequencing하므로 read들간의 overlap을 봄으로써 전체 sequence를 재구성하기에 더 용이하다는 장점이 있으며, 다만 비용적인 문제가 단점이라 할 수 있음.
+) 참고로 일반적으로 library를 만들고자 할 때는 한 read의 길이를 보통 200-300bp정도로 맞춰줌.
이제 이와 관련된 몇가지 실제 예시에 대해 생각해보자. 일단, 3T sequencing output을 가지는 sequencing 장비가 있을 때, 30X coverage로 genome 정보를 얻는다는 가정 하에 얼마나 많은 genome을 analyze할 수 있을지에 대해 생각해보자.
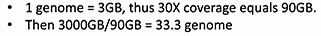
일단 1개의 genome은 3GB의 size이며, 이들이 30X coverage로 들어가므로 총 90GB에 해당함. 따라서 3T, 즉 3000GB를 90GB로 나누면 총 33.3개 종류의 genome을 분석할 수 있다는 것을 알 수 있음.
다음으로 3T sequencing output을 가지는 sequencing 장비를 가지고 얼마나 많은 read(fragment)를 얻을 수 있을지에 대해 생각해보자.

만약 150PE를 이용한다면, 이 경우 read는 2개이므로 300bp에 해당할 것임. 따라서 3T, 즉 3000Gbp를 300bp로 나누면 10,000,000,000개의 read를 얻을 수 있다는 것을 알 수 있음.
한편 100PE를 이용한다면 이 경우 3000Gbp를 200bp로 나눈 15,000,000,000개의 read만큼을 얻을 수 있음을 알 수 있음. 이를 봤을 때 150PE 대신 100PE를 이용하면 같은 output 성능을 가진 장비로 분석해도 1.5배나 더 많은 수의 read를 얻을 수 있다는 것을 알 수 있음. 실제로 RNA-seq, ChIP-seq과 같은 conventional data analysis를 수행할 때는 100PE만으로도 충분하므로, 100PE를 이용할 시 money를 아낄 수 있음.
다음으로 이제 DNA sequencing과 관련된 기본적인 principle들에 대해 알아보자.
기본적으로 DNA sequencing은 2가지 종류로 크게 나누어지는데, De novo sequencing과 resequencing이 바로 그것임. De novo sequencing은 지금까지 sequencing되지 않았던 unknown sequence를 최초로 sequencing해내는 것을 말하며, 이 sequencing을 수행할 때는 align을 위해 reference genome을 사용할 수 없음. (그렇기에 multiple technique을 교차적으로 이용하는 편이며, read 수를 많이 얻어내서 조합을 통해 신뢰할 만한 data를 얻어내야 함) 한편 resequencing은 align할 reference genome이 있는 상태로 수행하는 sequencing을 말하며, 이와 관련된 대표적인 sequencing example이 바로 ChIP-seq임.
DNA sequencing은 오랜 시간을 걸쳐 발전해왔는데, 우선 Sanger sequencing이 가장 고전적으로 많이 사용되었던 sequencing method임. 이 방법의 경우 capillary electrophoresis를 기반으로 해서 seqeuncing을 수행하며, 이후 많은 발전에 의해 automation과 추가적인 optimization이 이루어짐. 그럼에도 불구하고 이 방식으로 sequencing을 하게 될 시 시간이 너무 오래 소요되고 돈이 많이 소요된다는 단점이 있음. 참고로 1980년에 이러한 1st generation sequencing method를 개발한 공로로 Gilbert, Sanger 등의 과학자에게 노벨상이 수여됨. 한편 이후 cyclic method를 이용한 NGS(next generation sequencing) 기술이 개발됨.
다음 포스트부터는 1세대 시퀀싱 기술에 대해 알아보자.
'전공자를 위한 생물학 > 생물통계학(ChIP-seq)' 카테고리의 다른 글
| [생물통계학] 1.3 : next generation sequencing (NGS) - 1 (0) | 2025.05.20 |
|---|---|
| [생물통계학] 1.2 : 1세대 시퀀싱 (0) | 2025.05.17 |